

Geprüfte Informationen zu Krebs im Kindes- und Jugendalter in Kooperation mit Kinderkrebsinfo.de
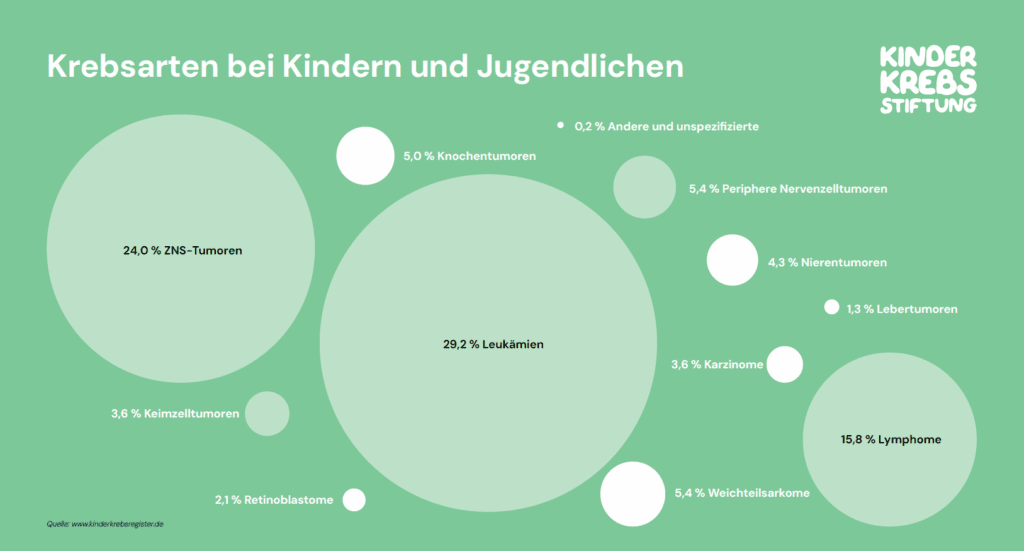
Bei bösartigen Erkrankungen im Kindes- und Jugendalter handelt es sich in den meisten Fällen um grundlegend andere Arten von Krebs als bei Erwachsenen. Prinzipiell kann jedes Gewebe im Körper entarten und jede gesunde Zelle kann sich zu einer Krebszelle entwickeln. So unterschiedlich wie die Gewebearten in unserem Körper sind, so vielgestaltig kann auch der Krebs sein. Während die meisten Tumoren im Erwachsenenalter der Gruppe der Karzinome zuzuordnen sind, sind bei Kindern bösartige Erkrankungen des Blutes am häufigsten. Leukämien machen etwa 30 % der Krebsneuerkrankungen aus. An zweiter Stelle stehen Tumoren des zentralen Nervensystems (ZNS – Gehirn und Rückenmark) mit 24 %, gefolgt von Krebserkrankungen des lymphatischen Gewebes mit etwa 15 %.

Krankheitsbilder, Ursachen, Diagnostik und Therapien: Auf über 3.000 Seiten in vier Sprachen bietet www.kinderkrebsinfo.de umfassende, wissenschaftlich fundierte und qualitätsgesicherte Informationen zu Blut- und Krebserkrankungen im Kindes- und Jugendalter – auch zu seltenen.
Eine Reihe von weiteren, relativ seltenen Krebs- und Bluterkrankungen werden – wie auch alle häufigeren Krebsarten bei Kindern und Jugendlichen – in Studien der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) untersucht. Einen Überblick über derzeit laufende Studien finden Sie auf der Übersichtsseite von Kinderkrebsinfo.de.
Seit 2009 wird das Informationsportal von der Deutschen Kinderkrebsstiftung gefördert. Die Informationen auf dieser Seite wurden mit freundlicher Genehmigung der Redaktion kinderkrebsinfo.de und der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) zur Verfügung gestellt.


Adenauerallee 134
53113 Bonn
Tel.: 02 28 / 68 84 6 – 0
Fax: 02 28 / 68 84 6 – 44
Deutsche Kinderkrebsstiftung
DE 04 3708 0040 0055 5666 16
DRESDEFF370
Commerzbank
USt.-IdNr.: DE 223 47 16 38

Sie sehen gerade einen Platzhalterinhalt von Instagram. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Google Maps. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Instagram. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr Informationen